



# 前言 #
歐盟體外診斷醫(yī)療器械新法規(guī)(IVDR) EU 2017/746 于2017年5月頒布,標志著98/79/EEC 廢止的五年過渡期開始。
2022年5月26日是IVDR的實施日期,標志著法規(guī)交替過渡期的結(jié)束。??
本文從IVDD指令到IVDR的最主要變化,到發(fā)證機構的現(xiàn)狀,歐盟政府的調(diào)整,以及廠家的應對幾方面進行梳理,希望對業(yè)內(nèi)的朋友有所啟發(fā)和幫助。
98/79/EEC IVDD 的“天生缺陷”
眾所周知,98/79/EEC的分類規(guī)則僅有寥寥數(shù)行,如下圖:

指令原文里面只有List A和 List B兩種分類,被囊括的產(chǎn)品更是極少,相比數(shù)量龐大的IVDD品類來說,這樣的分類只導致一種結(jié)果:
90%的IVD產(chǎn)品沒被納入進需要NB介入機構發(fā)證的范疇,也就是我們俗稱的others類產(chǎn)品。
EU 2017/746 IVDR的“大刀闊斧”
?IVDR在產(chǎn)品分類方面參照了2017/745 MDR 的分類規(guī)則,將IVD產(chǎn)品分成了Class A,B,C,D 4類,把具體的一類產(chǎn)品歸在一個分類規(guī)則下,比如:
檢測是否存在或顯露傳染性因子, 其會導致危及生命的疾病, 并且具有高的或可疑的傳播風險。?
這類產(chǎn)品,就屬于IVDR 風險最 高的Class D類產(chǎn)品。
IVDR的分類規(guī)則不再像IVDD指令那樣的劃分具體的產(chǎn)品個體,比如乙肝產(chǎn)品就劃分為List A類產(chǎn)品。
NB機構的“力不從心”
在近幾年的時間里,大量的IVD廠家的產(chǎn)品需要尋求發(fā)證機構的CE審核及發(fā)證服務,但是發(fā)證機構本身IVD專家資源的匱乏,IVDR發(fā)證機構的稀缺(到目前為止尚只有7家已經(jīng)獲取IVDR資質(zhì)的NB機構),加之整個IVD行業(yè)由于疫情的井噴式增長以及新法規(guī)的實施的疊加效應,導致了NB機構的“力不從心”。
歐盟政府的“被逼無奈”
作為歐洲NB機構的監(jiān)管方,歐盟政府就NB機構運力不足的問題,在2022年1月25日,由歐洲議會和歐盟委員會發(fā)布REGULATION (EU)2022/112,確定了IVDR 過渡期延期事宜。
? 依據(jù)發(fā)布的REGULATION (EU) 2022/112,具體延期如下:
-過渡期總體由 2024.5.27 推遲到 2025.5.27;
-在2022.5.26之前完成了NB發(fā)證的IVDD下List A, List B和Self-test產(chǎn)品,可繼續(xù)銷售至2025.5.26;
-對于 IVDD 下 other 類產(chǎn)品,在 2022.5.26 之前完成了IVDD符合性聲明(歐盟注冊)的,在如下日期之前仍可上市銷售:
? Class D(IVDR下)2025.5.26;
? Class C(IVDR下)2026.5.26;
? Class B(IVDR下)2027.5.26;
? Class A無菌(IVDR下)2027.5.26。
-對于 IVDD 下 other 類產(chǎn)品,在 2022.5.26 之后完成IVDD符合性聲明(歐盟注冊)的,在如下日期之前可上市銷售:
? Class D(IVDR下)2026.5.26;
? Class C(IVDR下)2027.5.26;
? Class B(IVDR下)2028.5.26;
? Class A無菌(IVDR下)2028.5.26。
? ?? ?IVDR 新政并沒有推遲,還是 2022.5.26 實施;PMS、警戒系統(tǒng)、經(jīng)濟運營商注冊這些要求,依舊自2022.5.26立即實施;由此,企業(yè)需及時更新QMS,同時依據(jù)IVDR要求實施企業(yè)注冊、產(chǎn)品注冊等。
IVD廠家的“未雨綢繆”
? ? ? ?很多IVD廠家前期在聯(lián)系NB發(fā)證機構之時可能都會被告知機構不受理,但是發(fā)證機構受理與否永遠是一個動態(tài)的不可預知的過程,IVDR產(chǎn)品眾多,相應的性能檢測,可用性檢測,臨床實驗都會花費較長的時間,IVD廠家比較恰當?shù)淖龇ň褪窃诘却龣C構的同時把相應的前置條件先完成,這樣,一旦發(fā)證機構受理,就可以趕上頭班車,大大節(jié)約等待和準備的時間。
已獲IVDR授權的公告機構,共計7家 (信息來源:NANDO)

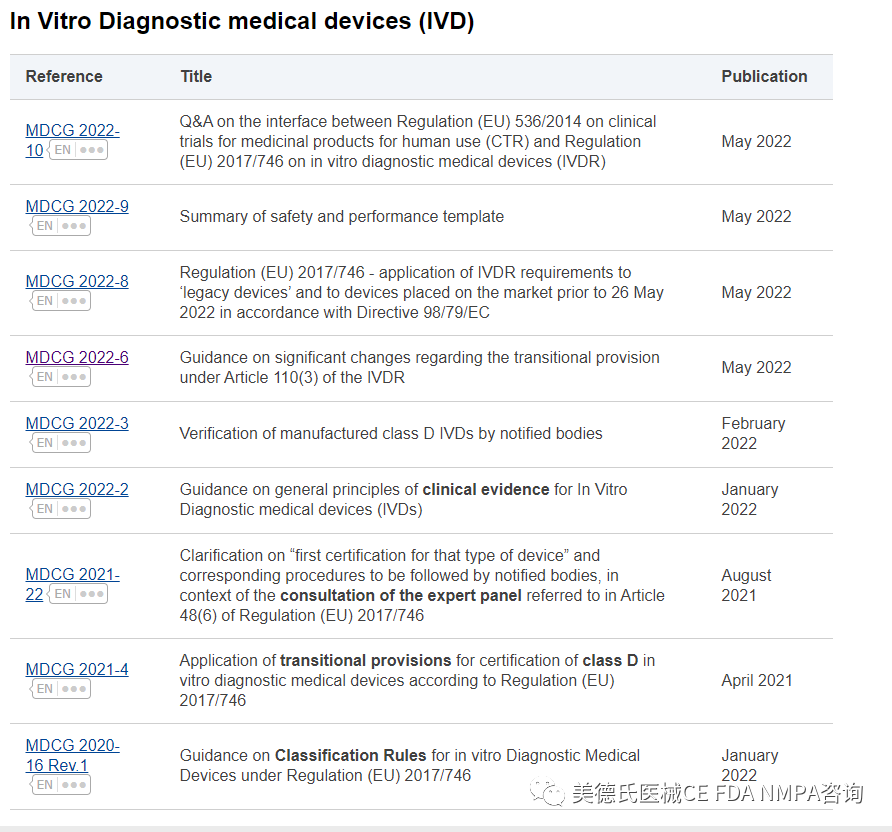
已發(fā)布的MDCG IVDR指南文件
本文相關鏈接
延期法案:
https://eur-lex.europa.eu/legal-content/EN/TXT/HTML/?uri=CELEX:32022R0112&from=EN
MDCG指南下載鏈接:
https://ec.europa.eu/health/md_sector/new_regulations/guidance_en
NANDO公告機構數(shù)據(jù)庫:
https://ec.europa.eu/growth/tools-databases/nando/
美德氏醫(yī)械服務范圍
質(zhì)量管理體系服務
服務 | 內(nèi)容 |
| ISO13485 | ?ISO ?13485的質(zhì)量體系的培訓、建立及運行輔導 |
| FDA | ?FDA CFR820?的質(zhì)量體系的培訓、建立及運行輔導 |
| MDSAP | ?MDSAP的質(zhì)量體系的培訓、建立及運行輔導 |
| NMPA | ?NMPA(GMP、GSP)的質(zhì)量體系的培訓、建立及運行輔導 |
| 質(zhì)量體系日常維護服務 | FDA820的不符合項以及警告信、歐盟的CAPA、流程改進、質(zhì)量體系維護的外包、供應商審核 |
法規(guī)合規(guī)及產(chǎn)品注冊服務
| 服務 | 內(nèi)容 |
| 歐盟市場準入整體解決方案 | 包括CE技術文檔撰寫、輔導、測試、認證全套方案。還包括歐代服務、歐盟FSC、ISO14971 風險分析、臨床評價、滅菌、軟件周期、可用性等歐盟合規(guī)的咨詢與服務 |
| 美國市場準入整體解決方案 | 包括510K文檔撰寫與認證,產(chǎn)品列名、工廠注冊、美國代理人、UDI的合規(guī)咨詢與服務 |
| 中國市場準入整體解決方案 | 包括NMPA文檔撰寫與注冊,生產(chǎn)許可證、中國FSC、醫(yī)療器械廣告審核的合規(guī)咨詢與服務 |
| 其他國家的認證注冊咨詢服務 | 包括全球法規(guī)注冊咨詢服務,如澳大利亞、新西蘭、加拿大、巴西、俄羅斯、日本、韓國等全球國家的注冊咨詢服務 |
醫(yī)療器械法規(guī)培訓
服務 | 內(nèi)容 |
| 國內(nèi)外法規(guī)培訓精講 | MDR 2017/745/EU?法規(guī)培訓 |
| IVDR 2017/746/EU?法規(guī)培訓 | |
| MDR臨床實驗方案設計培訓 | |
| IVDR臨床實驗,性能實驗方案設計培訓 | |
| ISO14971-2019?醫(yī)療器械風險管理培訓 | |
| 《ISO13485-2016醫(yī)療器械 質(zhì)量管理體系用于法規(guī)的要求》深度解讀 | |
| 產(chǎn)品設計開發(fā),產(chǎn)品可用性工程培訓 | |
| ISO 11135 11137 滅菌培訓 | |
| 其他定制式企業(yè)培訓 |