




法規(guī)框架
在美國(guó)銷(xiāo)售的醫(yī)療器械應(yīng)遵守《聯(lián)邦食品藥品和化妝品法案》(FD&C Act)的法規(guī)控制以及《標(biāo)題21-聯(lián)邦法規(guī)法典》(21 CFR)第1-58、800-1299部分的法規(guī) 。

美國(guó)醫(yī)療器械的法規(guī)歷史
1976: Medical Device Amendments to the FD&C Act
-旨在確保醫(yī)療器械的安全性和有效性;
-建立了一個(gè)基于產(chǎn)品風(fēng)險(xiǎn)的三級(jí)分類(lèi)系統(tǒng);
-建立了新醫(yī)療器械的上市法規(guī)路徑:上市前審批PMA和上市前通知510(k);
-建立了新的研究類(lèi)醫(yī)療器械(IDE)的法規(guī)路徑;
-建立了后市場(chǎng)的要求,包括場(chǎng)地注冊(cè),產(chǎn)品列名,不良事件的匯報(bào),GMP等;
1990: Safe Medical Devices Act (SMDA)
-加強(qiáng)后市場(chǎng)的監(jiān)管;
-定義了510(k) 中實(shí)質(zhì)性等同的含義。
1997: Food and Drug Administration Modernization Act (FDAMA)
-創(chuàng)建了第三方認(rèn)證的選項(xiàng),可以對(duì)某些設(shè)備進(jìn)行上市前的初步審查
-建立了De Novo計(jì)劃,通過(guò)該計(jì)劃,新型中低風(fēng)險(xiǎn)設(shè)備可以分類(lèi)為I級(jí)或II級(jí),而不是自動(dòng)將其分類(lèi)為III級(jí)
2002: Medical Device User Fee and Modernization Act (MDUFMA)
-授予FDA收取針對(duì)某些醫(yī)療器械上市前提交的使用費(fèi),以幫助FDA提高醫(yī)療器械提交審查的效率,質(zhì)量和可預(yù)測(cè)性
-實(shí)施了小型企業(yè)確定(SBD)計(jì)劃,以降低合格的小型企業(yè)的上市前批準(zhǔn)費(fèi)用
-為某些上市前提交的決定制定FDA績(jī)效目標(biāo)
-建立了針對(duì)“再加工”設(shè)備的新法規(guī)要求
-醫(yī)療器械公司的授權(quán)電子注冊(cè)
2007: Food and Drug Administration Amendments Act (FDAAA)
-重新修訂了醫(yī)療器械使用費(fèi),包括縮短了上市前審查時(shí)間;
-要求所有注冊(cè)和列名均以電子方式執(zhí)行;
-要求FDA為醫(yī)療設(shè)備建立的設(shè)備識(shí)別(UDI)系統(tǒng),以要求設(shè)備標(biāo)簽帶有的標(biāo)識(shí)符。
2016: 21st Century Cures Act
-通過(guò)定義不能作為設(shè)備進(jìn)行監(jiān)管的醫(yī)療軟件類(lèi)別,闡明了如何監(jiān)管某些數(shù)字保健產(chǎn)品。
2017: Food and Drug Administration Reauthorization Act (FDARA)
-重新修訂了醫(yī)療器械使用費(fèi)計(jì)劃;
-授權(quán)了針對(duì)設(shè)備企業(yè)的基于風(fēng)險(xiǎn)的檢查計(jì)劃,并規(guī)定了與設(shè)備企業(yè)審核相關(guān)的其他流程改進(jìn)。
FDA對(duì)于醫(yī)療器械的定義
設(shè)備,儀器,工具,機(jī)器,配件,植入物,體外試劑或其他類(lèi)似或相關(guān)的物品,包括以下組成部分或附件:
在官方國(guó)家處方或美國(guó)藥典或其任何補(bǔ)充中認(rèn)可的藥物,
- 旨在用于人類(lèi)或其他動(dòng)物的疾病或其他狀況的診斷或治愈,緩解,治療或預(yù)防疾病的用途,或
- 旨在影響人類(lèi)或其他動(dòng)物的身體的結(jié)構(gòu)或任何功能,并且無(wú)法通過(guò)人類(lèi)或其他動(dòng)物的身體內(nèi)部或之上的化學(xué)作用實(shí)現(xiàn)其主要預(yù)期目的,并且
它不能通過(guò)在人或其他動(dòng)物體內(nèi)或?qū)θ梭w的化學(xué)作用來(lái)達(dá)到其主要預(yù)期目的,并且不依賴(lài)于通過(guò)代謝來(lái)實(shí)現(xiàn)其主要預(yù)期目的。
美國(guó)醫(yī)療器械的主管機(jī)構(gòu)
美國(guó)食品藥品監(jiān)督管理局(FDA)對(duì)食品和藥品的監(jiān)督始于1906年,羅斯福總統(tǒng)簽署了《聯(lián)邦食品和藥品法》。從那時(shí)起,國(guó)會(huì)擴(kuò)大了FDA在保護(hù)和促進(jìn)人類(lèi)和獸藥,生物產(chǎn)品,醫(yī)療設(shè)備和輻射發(fā)射產(chǎn)品,人類(lèi)和動(dòng)物食品以及化妝品中的作用。醫(yī)療器械的主管部門(mén)為美國(guó)食品和藥品管理局(FDA)下屬的器械和放射健康中心(CDRH)。

醫(yī)療器械的上市路徑

-確定器械分類(lèi)
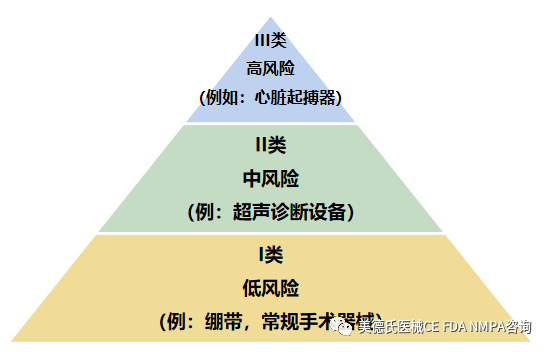
美國(guó)食品藥品監(jiān)督管理局(FDA)已為大約1,700種不同通用類(lèi)型的設(shè)備建立了分類(lèi),設(shè)備分類(lèi)是基于風(fēng)險(xiǎn)的,也就是說(shuō),設(shè)備對(duì)患者和/或用戶(hù)造成的風(fēng)險(xiǎn)是分配給設(shè)備的類(lèi)別中的主要因素。I類(lèi)為風(fēng)險(xiǎn)低的設(shè)備,III類(lèi)為風(fēng)險(xiǎn)大的設(shè)備。
-如何尋找器械分類(lèi)?
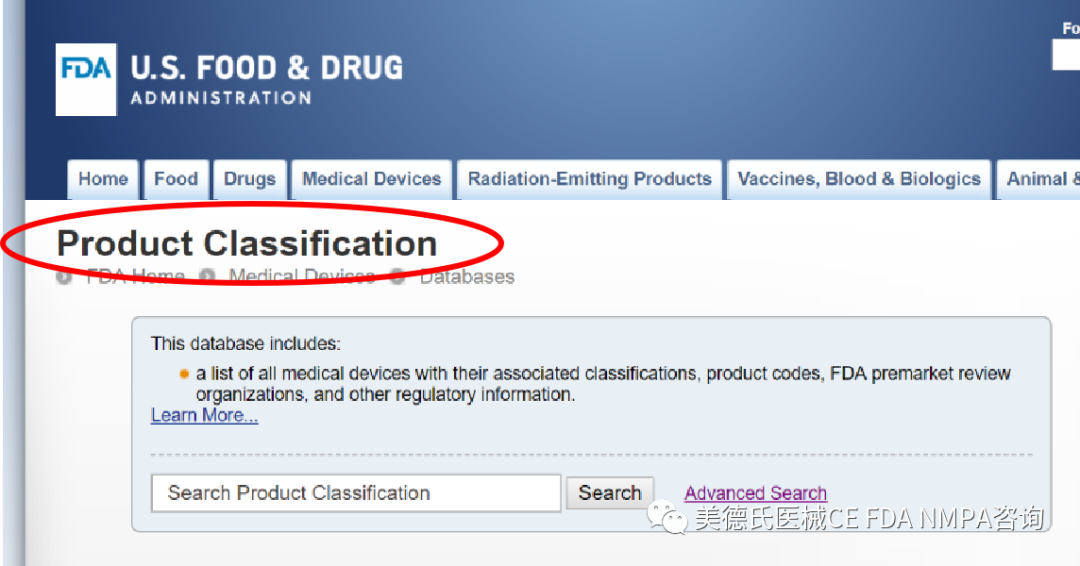
-選擇合適的上市前路徑
在對(duì)設(shè)備進(jìn)行分類(lèi)之后,選擇該法規(guī)所需的上市前路徑提交文件。上市前常見(jiàn)的提交方式包括:
510(k)(上市前通知)
PMA(上市前批準(zhǔn))
De Novo(自動(dòng)III級(jí)的評(píng)估)
HDE(人道主義設(shè)備豁免)
-510(k)(上市前通知)
510(k)是在醫(yī)療器械上市前遞交給FDA的一套文件,來(lái)證明所申請(qǐng)的產(chǎn)品與已在美國(guó)上市的產(chǎn)品是實(shí)質(zhì)等同(SE)的。申請(qǐng)人在拿到FDA的批準(zhǔn)信之前,該醫(yī)療器械是不能在美國(guó)上市的。
某些I類(lèi)設(shè)備不受上市前通知和/或良好生產(chǎn)規(guī)范的約束。大約572或74%的I類(lèi)設(shè)備免于上市前通知流程。
大部分II類(lèi)產(chǎn)品和部分I類(lèi)產(chǎn)品是需要通過(guò)510(k)的。
-實(shí)質(zhì)等同 Substantial Equivalence
實(shí)質(zhì)等同指的是該申請(qǐng)的產(chǎn)品與已上市產(chǎn)品具有同樣的安全性和有效性,即:
- 具有相同的預(yù)期用途;且具有相同的技術(shù)參數(shù);
或
具有相同的預(yù)期用途; 且與之對(duì)比后技術(shù)參數(shù)的不同沒(méi)有產(chǎn)生新的安全性和有效性的問(wèn)題;且遞交給FDA的資料可以證明此產(chǎn)品至少和已上市產(chǎn)品具有相同的安全性和有效性。
實(shí)質(zhì)等同的聲明不需要證明申請(qǐng)產(chǎn)品和對(duì)照產(chǎn)品是完全等同的,而可以通過(guò)對(duì)比預(yù)期用途,設(shè)計(jì),作用機(jī)理,材料,生產(chǎn)過(guò)程,性能等參數(shù)來(lái)建立。
-PMA (上市前批準(zhǔn))
大多數(shù)III類(lèi)設(shè)備需要PMA。PMA是嚴(yán)格的上市前提交類(lèi)型。在FDA批準(zhǔn)PMA之前,申辦者必須提供有效的科學(xué)證據(jù),以證明對(duì)設(shè)備預(yù)期用途的安全性和有效性的合理保證。
PMA通常需要提供臨床試驗(yàn)的資料,并且在審批前需要經(jīng)過(guò)FDA的現(xiàn)場(chǎng)審核。
-De Novo (上市前批準(zhǔn))
De Novo提供了一種在沒(méi)有有效的等同產(chǎn)品的情況下將符合條件的新設(shè)備分類(lèi)為I類(lèi)或II類(lèi)的方法。
-HDE (人道主義設(shè)備豁免)
HDE為旨在使患有罕見(jiàn)疾病或病癥的患者受益的III類(lèi)設(shè)備提供了一條監(jiān)管途徑。為了使設(shè)備符合HDE的資格,申辦者必須獲得人道主義使用設(shè)備(HUD)的指定,該名稱(chēng)是通過(guò)向FDA的孤兒產(chǎn)品開(kāi)發(fā)辦公室(OOPD)申請(qǐng)而獲得的。
-上市前提交的信息
根據(jù)選擇的不同的上市前申請(qǐng)的路徑,準(zhǔn)備相應(yīng)的資料包括:
產(chǎn)品描述:
型號(hào)規(guī)格
工作原理
結(jié)構(gòu)組成
技術(shù)指標(biāo)
預(yù)期用途
臨床前評(píng)價(jià)
臨床資料
標(biāo)簽,說(shuō)明書(shū)臨床資料
……
注:提供測(cè)試報(bào)告的實(shí)驗(yàn)室需要符合GLP的認(rèn)證
-FDA年費(fèi)及審核費(fèi)
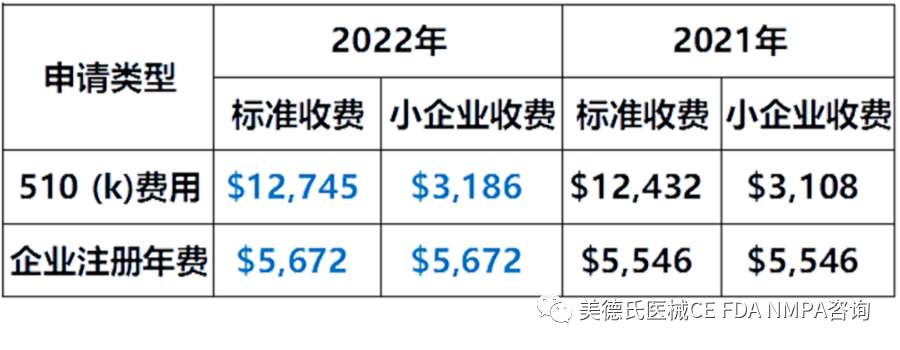
-eCopy 電子副本
上市前提交的內(nèi)容必須包含電子副本(eCopy),可以以光盤(pán)(CD),數(shù)字視頻光盤(pán)(DVD)或閃存驅(qū)動(dòng)器作為電子副本的存儲(chǔ)媒介。

-510(k)審核時(shí)間流程圖

上市后的監(jiān)管
-21 CFR Part 820 C Quality System Regulation
制造商必須建立并遵循質(zhì)量體系,以幫助確保其產(chǎn)品始終符合適用的要求和規(guī)格。FDA管制產(chǎn)品(食品,藥品,生物制品和設(shè)備)的質(zhì)量體系被稱(chēng)為當(dāng)前的良好生產(chǎn)規(guī)范(CGMP)。聯(lián)邦食品,藥品和化妝品法案首先授權(quán)了820部分(21 CFR 820部分)中設(shè)備的CGMP要求。該規(guī)定于1978年12月18日生效,并根據(jù)第820部分進(jìn)行了編纂。
QS法規(guī)適用于打算商業(yè)銷(xiāo)售醫(yī)療器械的成品器械制造商。
FDA已確定某些類(lèi)型的醫(yī)療設(shè)備不受GMP要求。這些設(shè)備不受《聯(lián)邦公報(bào)》中公布并在21 CFR 862至892中編纂的FDA分類(lèi)法規(guī)的豁免。免除GMP要求并不免除成品設(shè)備制造商保留投訴文件(21 CFR 820.198)或有關(guān)記錄保存的一般要求( 21 CFR 820.180)。

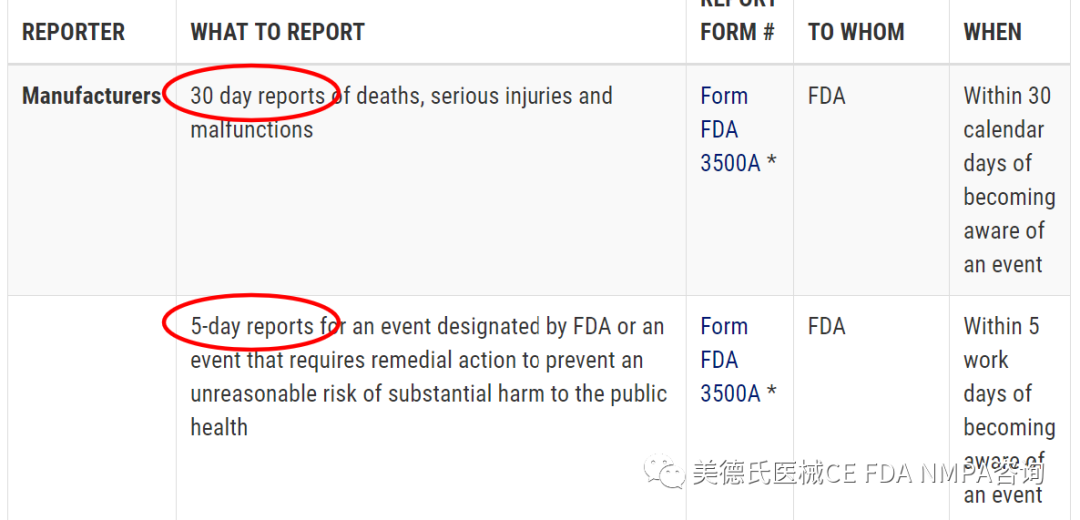
-醫(yī)療器械事故報(bào)告
醫(yī)療設(shè)備報(bào)告(MDR)法規(guī)(21 CFR第803部分)包含對(duì)制造商,設(shè)備用戶(hù)的強(qiáng)制性要求,以便向FDA報(bào)告某些與設(shè)備有關(guān)的不良事件和產(chǎn)品問(wèn)題。該法規(guī)規(guī)定,報(bào)告應(yīng)以FDA的Medwatch表格3500A或電子等效文件形式歸檔。FDA在2014年2月14日發(fā)布了規(guī)則,要求制造商以電子格式向FDA提交MDR,以便FDA可以對(duì)其進(jìn)行處理,審查和存檔。該規(guī)定將于2015年8月14日生效。

-匯報(bào)時(shí)限
- 醫(yī)療器械的糾正和移除
根據(jù)21 CFR 806,醫(yī)療器械的糾正和移除,制造商必須向FDA報(bào)告醫(yī)療器械的任何糾正或移除,前提是該糾正或移除是為了減少醫(yī)療器械引起的健康風(fēng)險(xiǎn)。或糾正由該設(shè)備引起的違反本法的行為,而該行為可能會(huì)危害健康。

-UDI
UDI規(guī)則要求設(shè)備標(biāo)簽商(通常是制造商)必須:
在設(shè)備標(biāo)簽,設(shè)備包裝上,有時(shí)在設(shè)備上直接包含在FDA認(rèn)可的發(fā)行機(jī)構(gòu)的UDI系統(tǒng)下發(fā)布的設(shè)備標(biāo)識(shí)符(UDI)。
將設(shè)備信息提交到設(shè)備標(biāo)識(shí)數(shù)據(jù)庫(kù)(GUDID)。

總結(jié)
選擇合適的上市路徑,提交合適的文件
考慮多個(gè)上市地的要求,提前規(guī)劃,節(jié)約資源
和FDA及時(shí)溝通,及時(shí)獲得反饋
注意盡早的符合體系相關(guān)的要求
美德氏醫(yī)械服務(wù)范圍
質(zhì)量管理體系服務(wù)
| 服務(wù) | 內(nèi)容 |
| ISO13485 | ?ISO?13485的質(zhì)量體系的培訓(xùn)、建立及運(yùn)行輔導(dǎo) |
| FDA | ?FDA CFR820?的質(zhì)量體系的培訓(xùn)、建立及運(yùn)行輔導(dǎo) |
| MDSAP | ?MDSAP的質(zhì)量體系的培訓(xùn)、建立及運(yùn)行輔導(dǎo) |
| NMPA | ?NMPA(GMP、GSP)的質(zhì)量體系的培訓(xùn)、建立及運(yùn)行輔導(dǎo) |
| 質(zhì)量體系日常維護(hù)服務(wù) | FDA820的不符合項(xiàng)以及警告信、歐盟的CAPA、流程改進(jìn)、質(zhì)量體系維護(hù)的外包、供應(yīng)商審核 |
| 服務(wù) | 內(nèi)容 |
| 歐盟市場(chǎng)準(zhǔn)入整體解決方案 | 包括CE技術(shù)文檔撰寫(xiě)、輔導(dǎo)、測(cè)試、認(rèn)證全套方案。還包括歐代服務(wù)、歐盟FSC、ISO14971 風(fēng)險(xiǎn)分析、臨床評(píng)價(jià)、滅菌、軟件周期、可用性等歐盟合規(guī)的咨詢(xún)與服務(wù) |
| 美國(guó)市場(chǎng)準(zhǔn)入整體解決方案 | 包括510K文檔撰寫(xiě)與認(rèn)證,產(chǎn)品列名、工廠注冊(cè)、美國(guó)代理人、UDI的合規(guī)咨詢(xún)與服務(wù) |
| 中國(guó)市場(chǎng)準(zhǔn)入整體解決方案 | 包括NMPA文檔撰寫(xiě)與注冊(cè),生產(chǎn)許可證、中國(guó)FSC、醫(yī)療器械廣告審核的合規(guī)咨詢(xún)與服務(wù) |
| 其他國(guó)家的認(rèn)證注冊(cè)咨詢(xún)服務(wù) | 包括全球法規(guī)注冊(cè)咨詢(xún)服務(wù),如澳大利亞、新西蘭、加拿大、巴西、俄羅斯、日本、韓國(guó)等全球國(guó)家的注冊(cè)咨詢(xún)服務(wù) |
| 服務(wù) | 內(nèi)容 |
| 國(guó)內(nèi)外法規(guī)培訓(xùn)精講 | MDR 2017/745/EU?法規(guī)培訓(xùn) |
| IVDR 2017/746/EU?法規(guī)培訓(xùn) | |
| MDR臨床實(shí)驗(yàn)方案設(shè)計(jì)培訓(xùn) | |
| IVDR臨床實(shí)驗(yàn),性能實(shí)驗(yàn)方案設(shè)計(jì)培訓(xùn) | |
| ISO14971-2019?醫(yī)療器械風(fēng)險(xiǎn)管理培訓(xùn) | |
| 《ISO13485-2016醫(yī)療器械 質(zhì)量管理體系用于法規(guī)的要求》深度解讀 | |
| 產(chǎn)品設(shè)計(jì)開(kāi)發(fā),產(chǎn)品可用性工程培訓(xùn) | |
| ISO 11135 11137 滅菌培訓(xùn) | |
| 其他定制式企業(yè)培訓(xùn) |